What is Sickle Cell Disease?
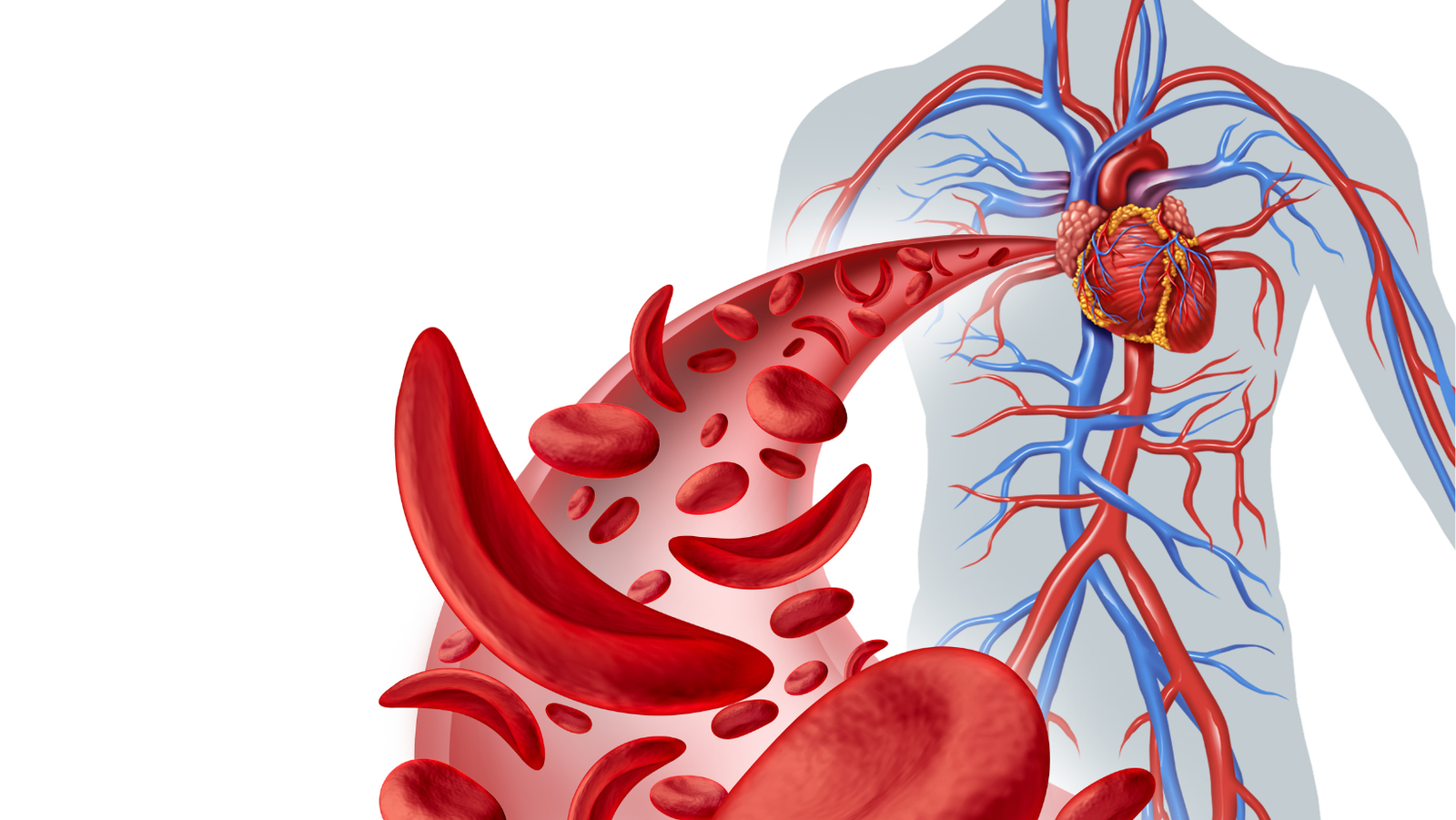
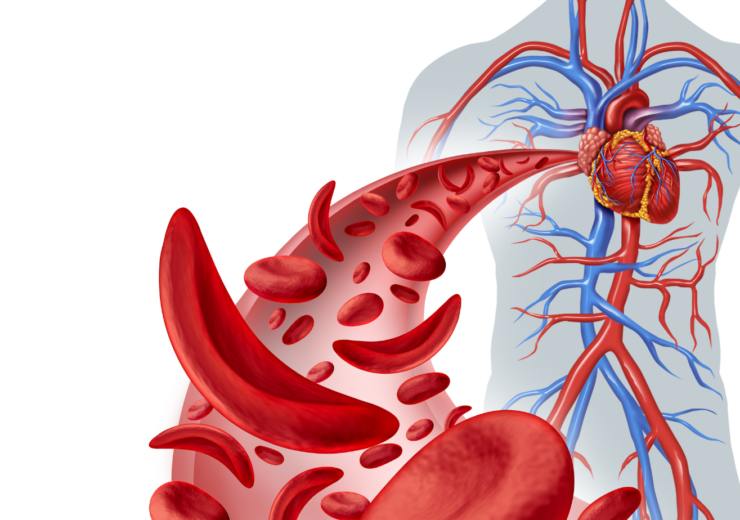
Sickle Cell Anemia is an inherited blood disease that has a genetic defect in the hemoglobin. Hemoglobin is a protein that all red blood cells contain, and oxygen is carried by hemoglobin from the lungs to all parts of the body.
Red blood cells that contain normal hemoglobin are soft and round. Their soft texture enables them to squeeze through the body’s small blood vessels.
People with Sickle Cell Anemia, however, have a type of abnormal hemoglobin called hemoglobin S. (Normal hemoglobin is called hemoglobin A). A genetic error makes the hemoglobin molecules stick together in long, rigid rods after they release oxygen. These rods cause the red blood cells to become hard and sickle-shaped, unable to squeeze through tiny blood vessels. The misshapen cells can get stuck in the small blood, causing a blockage that deprives the body’s cells and tissues of blood and oxygen.
When this happens it’s like having mini heart attacks throughout the entire body. A heart attack is painful because the blood flow to the heart is interrupted. In Sickle-Cell Anemia, the blood flow can be interrupted to any of the major organs, causing severe pain and organ damage at the site of the blood flow blockage
.
These painful “crises”, as they are called, damage the lungs, kidneys, liver, bones, and other organs and tissues. Recurrence of these episodes is the most disabling feature of Sickle-Cell Anemia. They can cause leg ulcers, blindness, and many other health problems, depending upon where in the body the blood flow blockage occurs. A blockage in the brain can cause a stroke, which may result in paralysis or death.
The body, recognizing that the sickle cells are abnormal, destroys them at a faster rate than it can replace them. This causes a type of anemia, a shortage of red blood cells. Symptoms of anemia include extreme fatigue and susceptibility to infection.
There are approximately 250 million (250,000,000) sufferers of hemoglobinopathies such as Sickle-Cell Anemia worldwide, and 300,000 infants are born each year with the disease. Sickle cell hemoglobinopathies are also found in Caucasian, Native Americans, Hispanics, Asian, Japanese, Italian, Chinese, Arabian and Israelis.
People whose ancestors came from parts of the world where malaria was prevalent are potential carriers of the sickle gene.
In the past, individuals with Sickle-Cell Anemia often died in childhood. A 1973 study estimated that half of those with the disease died by the age of 14. A frequent cause of death was bacterial infection by the organism Streptococcus pneumoniae. Children with Sickle-Cell Anemia are highly susceptible to infections because they lose the function of their spleen, which provides protection against bacterial infections.
Rates of early death are highest among those with a severe form of the disease. Between 10 and 15 percent of patients will have three or more painful crises per year. The more crises you have per year, the greater your chances of dying prematurely. Your organs become more damaged when they chronically are not receiving enough blood and oxygen.
The concept of the Orthomolecular treatment of diseases was first defined by two-time Nobel Laureate, Dr. Linus Pauling:
I believe that in general the treatment of disease by the use of substances, such as ascorbic acid, that are normally present in the human body and are required for life is to be preferred to treatment by the use of powerful synthetic substances or plant products, which may, and usually do, have undesirable side effects. Such substances as Vitamin C and most of the other vitamins are remarkable for their low toxicity and absence of side effects when taken in amounts larger than those usually available in the diet. I have coined the term Orthomolecular medicine for the preservation of good health and the treatment of disease by varying the concentrations in the human body of substances that are normally present in the body and are required for health.
Death by starvation, kwashiorkor, beriberi, scurvy, or any other deficiency disease can be averted by providing an adequate daily intake of carbohydrates, essential fats, proteins (including the essential amino acids), essential minerals, and thiamine, ascorbic acid, and other vitamins. To achieve the best of health, the rate of intake of essential foods should be such as to establish and maintain the optimum concentrations of essentials molecules, such as those of ascorbic acid. (Pauling, 1968 B)